
 W4M : http://workflow4metabolomics.org/
W4M : http://workflow4metabolomics.org/
Workflow4Metabolomics est un portail collaboratif dédié au traitement, à l’analyse et à l’annotation des données métabolomiques.L’Institut Français de Bioinformatique (IFB) et l’infrastructure MetaboHUB ont développé des pipelines complets de LC / MS, GC / MS et de RMN en utilisant le cadre de Galaxy pour l'analyse des données, y compris le prétraitement, la normalisation, le contrôle de la qualité, l'analyse statistique (univariée, multivariée PLS / OPLS) et les étapes d'annotation.
 PeakForest : https://peakforest.org/
PeakForest : https://peakforest.org/
La base de données MetaboHUB PeakForest fournit des services de stockage et d'annotation pour les profils métaboliques des matrices biologiques et de référence. Grâce à son portail Web, PeakForest est consacré à l'annotation à haut débit et à l'identification de novo des métabolites. Elle repose sur la vaste gamme de méthodes complémentaires en utilisant l’UPLC- (API) HRMS, le GC-QToF et la RMN. Cette base de données est disponible dans les plateformes de MetaboHUB pour effectuer des analyses métabolomique non ciblées sur les biofluides (par exemple, le plasma humain et l'urine), des échantillons de tissus (par exemple, le fruit de la tomate) et des extraits cellulaires (par exemple, E. coli et S. cerevisiae). NMRProcFlow: http://www.nmrprocflow.org/
NMRProcFlow: http://www.nmrprocflow.org/
Le logiciel libre NMRProcFlow fournit un ensemble complet d'outils pour le traitement et la visualisation des données 1D RMN, le tout au sein d'une interface interactive basée sur une visualisation des spectres. BiotStatFlow : http://biostatflow.org/
BiotStatFlow : http://biostatflow.org/
BioStatFlow facilite l'accès à des outils statistiques pour les biologistes qui ne sont pas des spécialistes. Il a été conçu pour exécuter des analyses statistiques séquentiellement, à savoir une chaîne linéaire de l'analyse statistique, que l'on appelle Workflow pour les données «omiques». MetExplore : www.metexplore.fr
MetExplore : www.metexplore.fr
MetExplore est un site Web qui offre plusieurs fonctionnalités pour l’analyse des réseaux métaboliques :
MS-CleanR: A package for cleaning and annotating LC-MS data
The MS-CleanR package provides functions for feature filtering and annotation of LC-MS data.
See the publication and tutorials (pdf files included in the master branch) for more information.
Needs MS-DIAL (v4.00 or higher) and MS-FINDER (3.30 or higher): http://prime.psc.riken.jp/compms/index.html
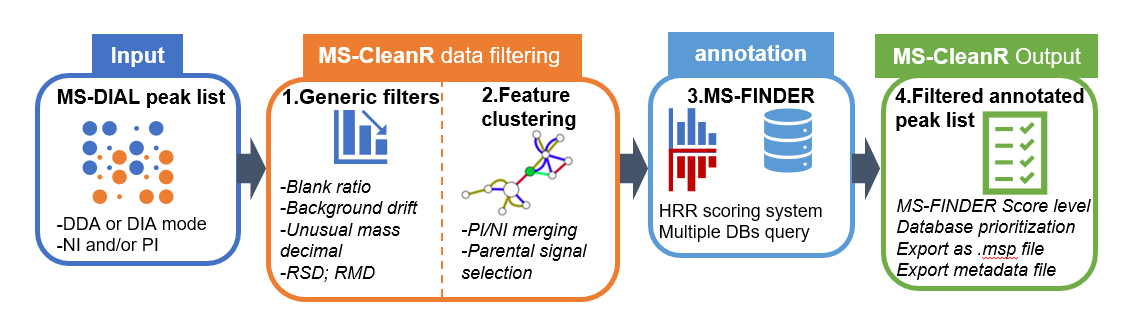
MS-CleanR use as input MS-DIAL peak list processed in data dependent analysis (DDA) or data independent analysis (DIA) using either positive ionization mode (PI) or negative ionization mode (NI) or both. First, MS-CleanR apply generic filters encompassing blank injection signal subtraction, background ions drift removal, unusual mass defect filtering, relative standard deviation threshold (RSD) based on sample class and relative mass defect (RMD) window filtering. All these options are tunable by the user. The second step involves a feature clustering method based on MS-DIAL peak character estimation algorithm followed by parental signal extraction using multi-level optimization of modularity algorithm. Optionally, MS-CleanR can merge PI and NI mode during this step. Then, all selected features are exported to MS-FINDER program for in silico-based annotation using hydrogen rearrangement rules (HRR) scoring system. At this step, multiple databases can be queried and each annotation results will be handled by MS-CleanR. The final step will merge annotation results to the filtered peak list by prioritizing database annotation depending on user choice. Optionally, all results can be exported as .msp file for mass spectral similarity networking purpose.
Installation
devtools::install_github("eMetaboHUB/MS-CleanR") library(mscleanr) runGUI()
Citation
Publication link: https://www.biorxiv.org/content/10.1101/2020.04.09.033308v2
Credits
Licence
GPL-3